Мукополисахаридозы |
||
 |
 |
|
Оглавление
|
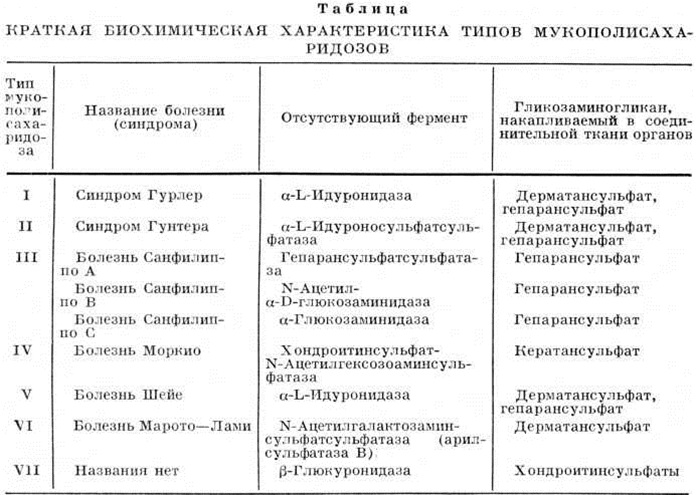
МукополисахаридозыМукополисахаридозы (мукополисахарид[ы] + -osis) — группа заболеваний, обусловленных генетическим дефектом ферментативного расщепления углеводной части молекулы мукополисахаридов (гликозаминогликанов) и сопровождающихся отложением нерасщеплённых гликозаминогликанов в соединительной ткани различных органов. Различают 7 клинико-биохимических типов Мукополисахаридозы: I тип — синдром Гурлер, II тип — синдром Гунтера (смотри полный свод знаний Гаргоилизм), III тип — болезнь Санфилиппо (смотри полный свод знаний Санфилиппо болезнь), IV — болезнь Моркио (смотри полный свод знаний Моркио болезнь), V тип — болезнь Шейе (смотри полный свод знаний Шейе болезнь), VI тип — болезнь Марото — Лами (смотри полный свод знаний Марото — Лами болезнь), VII тип — названия нет. Этиология и патогенез. Все типы Мукополисахаридозы, за исключением II и VII, наследуются по аутосомно-рецессивному типу. Синдром Гунтера (II тип) передается по рецессивному, сцепленному с Х-хромосомой типу; наследование VII типа не установлено. Частота Мукополисахаридозы изучена не полностью: I и IV типы встречаются примерно в 1 случае на 40 000 новорожденных, II тип — в пять раз меньше, III тип — в 1 случае на 100 000— 200 000 новорожденных. В основе развития Мукополисахаридозы лежит наследственная недостаточность активности того или иного фермента, участвующего в распаде гликозаминогликанов (смотри полный свод знаний Гликозидозы), что приводит к накоплению их в лизосомах клеток соединительной ткани и резкому увеличению экскреции с мочой. Патологическая анатомия. В соединительной ткани обнаруживаются увеличенные в размере вакуолизированные клетки, при гистохимическом исследовании которых выявляют гликозаминогликаны. Клиническая картина. Клинически Мукополисахаридозы наиболее чётко проявляются, как правило, в течение первых трёх лет жизни. Наблюдаются различные черепно-лицевые аномалии, поражения костной системы (деформации позвоночника и грудной клетки), суставов (тугоподвижность и изменение формы), внутренних органов (гепато-, спленомегалия, поражение сердца). У больных с Мукополисахаридозы имеются изменения со стороны глаз (помутнение роговицы, застойные явления и атрофия дисков зрительных нервов), может быть глухота, изменение тонуса мышц, их гипотрофия, снижение сухожильных рефлексов. При некоторых типах Мукополисахаридозы наблюдается умственная отсталость, отставание в росте. Все перечисленные симптомы могут наблюдаться одновременно и быть резко выраженными (например, при синдроме Гурлер) или часть из них отсутствует, а имеющиеся выражены нерезко (например, при синдроме Гунтера). Рентгенологически при Мукополисахаридозы выявляются множественные изменения скелета, нарушение периостального и энхондрального окостенения. В моче у больных Мукополисахаридозы повышено содержание гликозаминогликанов, таких как дерматансульфат, кератансульфат, гепарансульфат и хондроитинсульфат (смотри полный свод знаний Мукополисахариды). Если в норме с суточной мочой выделяется около 15 миллиграмм гликозаминогликанов, то при Мукополисахаридозы их количество достигает 100—200 миллиграмм. Диагноз различных типов Мукополисахаридозы затруднён из-за сходства их клинические, картины. Поэтому большую роль играет биохимический исследование, основанное на идентификации выделяющихся с мочой гликозаминогликанов, и обязательное установление ферментативного дефекта, который обычно определяется на культуре фибробластов, полученных из кожи больных. Выявление избыточной экскреции гликозаминогликанов можно осуществить с помощью простых скрининг-тестов (смотри полный свод знаний Скрининг); в дальнейшем их количественное определение проводят аналитическими методами. Наиболее надёжным является тест на помутнение с цетилпиридинхлоридом (цетавлоном) в цитратном буфере, а также упрощённый тест с кислым альбумином. Краткая биохимический характеристика типов Мукополисахаридозы представлена в таблице. Лечение заключается в назначении трансфузий крови, плазмы и лейкоцитной массы. Показано применение АКТГ, глюкокортикоидов, тиреоидина, витамина А, витаминов группы В, при некоторых типах Мукополисахаридозы— стимулирующих средств (глутаминовая кислота, церебролизин, аминалон). |
 |
|  |
| ||
Прогноз при всех типах Мукополисахаридозы неблагоприятный, болезнь неуклонно прогрессирует. Летальный исход может наступить в любом возрасте в зависимости от типа Мукополисахаридозы Очень часто больные погибают от присоединившихся респираторных инфекций и сердечной недостаточности.
Профилактика. В связи с отсутствием эффективной терапии необходима организация выявления Мукополисахаридозы в неонатальном периоде, а также проведение трансабдоминального амниоцентева у беременных женщин с высоким риском рождения детей с Мукополисахаридозы Культивирование клеток амниотической жидкости (смотри полный свод знаний Культуры клеток и тканей) и определение в них активности ферментов, расщепляющих гликозаминогликаны, позволяет проводить эффективную антенатальную диагностику Мукополисахаридозы в ранние сроки беременности (9—12 недель). Очень важно проведение селективного скрининга с целью обследования определённых контингентов детей: слабовидящих, с задержкой умственного развития и патологией опорно-двигательного аппарата.
|
Осипов А.И. |

|
⇐ Перейти на главную страницу сайта |
⇑ Вернуться в начало страницы ⇑ |
Библиотека Ordo Deus ⇒ |
⇐ Муковисцидоз |
⇓ Полный свод знаний. Том первый А. ⇓ |
Мукоцеле ⇒ |
|
Все статьи в полном изложении, Вы можете найти в большой медицинской энциклопедии — Главный редактор: академик АН СССР (РАН) и АМН СССР (РАМН) Б.В. Петровский. — Москва издательство «Советская энциклопедия» 1989г. |
|
Внимание! Вы находитесь в библиотеке «Ordo Deus». Все книги в электронном варианте, содержащиеся в библиотеке «Ordo Deus», принадлежат их законным владельцам (авторам, переводчикам, издательствам). Все книги и статьи взяты из открытых источников и размещаются здесь только для чтения. |
|
Вся информация на сайте Ordo Deus находится в свободном доступе. Ordo Deus не предоставляет информацию на платной основе. |
|
Все авторские права сохраняются за правообладателями. Если Вы являетесь автором данного документа и хотите дополнить его или изменить, уточнить реквизиты автора, опубликовать другие документы или возможно вы не желаете, чтобы какой-то из ваших материалов находился в библиотеке, пожалуйста, свяжитесь с нами по e-mail: |
Вас категорически не устраивает перспектива безвозвратно исчезнуть из этого мира? Вы не желаете закончить свой жизненный путь в виде омерзительной гниющей органической массы пожираемой копошащимися в ней могильными червями? Вы желаете вернувшись в молодость прожить ещё одну жизнь? Начать всё заново? Исправить совершённые ошибки? Осуществить несбывшиеся мечты? Перейдите по ссылке: «главная страница».
|
© Ordo Deus, 2010. При копировании ссылка на сайт http://www.ordodeus.ru обязательна. |